Определение дезоморфина в моче
/ Катаев С.С., Зеленина Н.Б., Шилова Е.А. — 2007.
Определение дезоморфина в моче / Катаев С.С., Зеленина Н.Б., Шилова Е.А. // Проблемы экспертизы в медицине, 2007, №1, с.32-36.
ОПРЕДЕЛЕНИЕ ДЕЗОМОРФИНА В МОЧЕ
С.С. Катаев, Н.Б. Зеленина, Е.А. Шилова
Пермское областное бюро судебно-медицинской экспертизы (нач. бюро – В.И. Перминов)
Приведены методы идентификации и количественного определения дезоморфина, а также морфина и кодеина в моче. Описаны хроматографические характеристики дезоморфина для хроматографии в тонком слое сорбента. Даны газо-хроматографические и масс-спектрметрические данные для дезоморфина и некоторых его производных, которые могут применяться для качественной идентификации последнего. Применение в скрининге дезоморфина иммунохроматографического анализа является более эффективным методом в сравнении с исследованием в тонком слое сорбента.
Ключевые слова: опиаты, дезоморфин, тонкослойная хроматография, иммуноанализ, газовая хроматография – масс-спектрометрия.
DETERMINATION DESOMORPHINE in URINE
S.S. Kataev, N.B. Zelenina, E.A. Shilova
The methods of identification and quantitative determination desomorphine with morphine and codeine in urine are broughted. The data of desomorphine for chromatography in thin layer of the sorbent are described. The data of gas chromatographic and mass-spectrometry for desomorphine and some its derived are given, which can be used for its qualitative identification. Using in screening of desomorphine by immunochromatographic assay is more efficient method in comparison with study in thin layer of the sorbent.
The keywords: opiates, desomorphine, thin-line chromatography, immunoassay, gas chromatography - mass-spectrometry.
Дезоморфин, синонимы: Dihydrodeoxymorphine, Desomorfin, Desomorphin(-e, -um), Desoximorfina, Dezomorfina, Dihidrodesoximorfina, Dihydrodesoksymorfin, Dihydrodesoximorfin, Dihydrodesoxymorfin, Dihydrodesoxymorphin(-e, -um), Dihydrodesoxymorphine-D, Escopermida, Permonid(-a), Scopermid. Химическое название: 4,5.альфа.-эпокси-17-метилморфинан-3-ол. Регистрационный номер CAS – 427-00-9.
Дезоморфин является наркотическим анальгетиком. Данное соединение в девять раз активнее морфина и в пять раз токсичней [3]. В России он включен в Список 1 Перечня наркотических и психотропных средств ПККН.
В ряде субъектов Приволжского региона фиксировалось употребление дезоморфина, синтезированного кустарным способом из препаратов, содержащих кодеин, что привело к необходимости определения дезоморфина в биологических объектах.
Оборудование
Газовый хроматограф Agilent 6850, оснащенный капиллярной кварцевой колонкой НР-5MS длиной 30 м с внутренним диаметром 0,25 мм и толщиной пленки 0,25 мкм. Масс-селективный детектор Agilent 5973N (Agilent, США). Термоблок ПЭ-4030 (ОАО “Экрос”, Россия). Полуавтоматические пипетки-дозаторы, позволяющие отбирать объемы жидкостей 4-40, 40-200, 200-1000 мкл и 1-5 мл.
Материалы и методы
Все используемые растворители и реактивы имели чистоту х.ч. Иммунохроматографические тесты для качественного экспресс-определения морфина и его метаболитов в моче NARCOSCREEN МОР (InTec Products Inc, Китай). Пластинки для тонкослойной хроматографии "Сорбфил-ПТСХ-В-УФ" (ЗАО "Сорбполимер", Россия). В качестве стандартов использовали этилморфина гидрохлорид (ГФ X, ФС 41), морфина гидрохлорид (ГФ X, ФС 413), кодеин основание (ГФ X, ФС 170). Дезоморфин выделяли из мочи субъектов, употреблявших наркотические средства, полученные кустарным способом. Очистку проводили с использованием тонкослойной хроматографии. Идентифицировали дезоморфин с помощью хромато-масс-спектрометрии. Количественное определение выделенного дезоморфина проводили методом газовой хроматографии с пламенно-ионизационным детектором по массовому коэффициенту с использованием в качестве внутреннего стандарта метилстеората. Анализировали образцы мочи лиц, подозреваемых в употреблении наркотических средств. Пробы мочи до исследования хранились при + 4оС.
Исследование проб мочи в тонком слое сорбента. К 10 мл мочи прибавляли 2 мл концентрированной хлористоводородной кислоты, флакон плотно закрывали и выдерживали 15 мин на кипящей водяной бане. После охлаждения гидролизат экстрагировали 10 мл хлороформа (экстракт отбрасывали). К водной фракции прибавляли 25% водный раствор аммиака до рН 9-10 и дважды экстрагировали смесью хлороформ – бутанол-1 (6:1) порциями по 10 мл. Органические фазы отделяли, объединяли и фильтровали через бумажный фильтр с безводным сульфатом натрия. Экстракт выпаривали до сухого остатка в токе теплого воздуха (60оС).
Сухой остаток извлечения из мочи растворяли в 50-100 мкл этанола и наносили поровну в стартовые зоны 1 двух хроматографических пластин, а в зоны 2 наносили спиртовые растворы метчиков морфина, кодеина и дезоморфина. Пластинки элюировали в системе растворителей толуол – ацетон – этанол – 25% водный раствор аммиака (45:45:7,5:2,5). Предварительное насыщение камеры 30 мин. Длина пробега фронта элюента 8 см. Хроматограммы высушивали. Первую пластинку проявляли реактивом Марки (капельно). Вторую – реактивом Фреде (капельно).
При наличии окрашенных зон на уровне пятен метчиков морфина (красно-фиолетовое пятно с hRf 20 ± 2) и/или кодеина (синее пятно с hRf 32 ± 5) и/или дезоморфина (красно-фиолетовое пятно с hRf 37 ± 5) фиксировали положительный результат.
Иммунохроматографический анализ мочи. Исследование проводили в соответствии с прилагаемой инструкцией. Во флакон помещали 2 мл мочи, контролировали рН по универсальной индикаторной бумаге, доводили температуру до комнатной. В мочу помещали тестовую полоску, через 30 секунд вынимали и помещали на ровную горизонтальную поверхность. Результат определяли в интервале 5-10 минут. При наличии одной окрашенной полосы в контрольной зоне фиксировали положительный результат. При наличии двух полос в контрольной и тестовой зонах фиксировали отрицательный результат.
Подготовка проб мочи для хромато-масс-спектрометрического исследования. 2 мл мочи помещали в стеклянный флакон объемом 10 мл, вносили 50 мкл спиртового раствора этилморфина гидрохлорида с концентрацией 0,02 мг/мл (внутренний стандарт) и добавляли 0,4 мл концентрированной хлористоводородной кислоты. Флакон плотно укупоривали и выдерживали в термоблоке при 100оС в течение 15 мин. Гидролизат охлаждали, экстрагировали хлороформом 2 раза порциями по 2 мл (экстракты отбрасывали). К водной фракции прибавляли 0,6 мл 25% водного раствора аммиака. Контролировали рН (9-10 по универсальной индикаторной бумаге) и дважды экстрагировали смесью хлороформ – бутанол-1 (6:1) порциями по 2 мл. Органические фазы отделяли, объединяли и фильтровали через бумажный фильтр с безводным сульфатом натрия. Экстракт выпаривали до сухого остатка в токе теплого воздуха (60оС). К сухому остатку прибавляли 40 мкл пиридина и 60 мкл уксусного ангидрида. Полученную смесь переносили в реакционную виалу на 2 мл, которую плотно укупоривали и выдерживали в термоблоке при 80оС в течение 30 мин. Избыток реагентов удаляли в токе теплого воздуха. Сухой остаток растворяли в 200 мкл безводного этилацетата и 1 мкл полученного раствора вводили в испаритель хромато-масс-спектрометра с использованием устройства для автоматического ввода проб. Режим работы автосамплера включал следующие операции: пять промывок шприца этанолом; пять промывок этилацетатом; одна промывка шприца раствором пробы; три прокачки раствором пробы перед вводом ее в хромато-масс-спектрометр; набор и ввод пробы; пять промывок шприца этанолом; пять промывок этилацетатом.
Режим работы газового хроматографа с масс-селективным детектором. Скорость потока газа-носителя (гелий) через колонку 1,5 мл/мин, режим работы split/splitless (деление потока 15:1, с задержкой включения 1 мин после ввода пробы). Температура испарителя хроматографа и интерфейса детектора задавалась 250 и 280оС, соответственно. Температура колонки начальная 70оС в течение 2 мин и прогрев до 280оС со скоростью программирования 20 град/мин, выдержка при конечной температуре 8 мин. Напряжение на умножителе масс-селективного детектора устанавливали на 200 В выше величины автоматической настройки детектора. Регистрация масс-спектров в режиме полного сканирования ионов в интервале масс 45-450 а.е.
Обработку хроматограмм с целью идентификации компонентов пробы проводили с использованием программы AMDIS (The Automatic Mass Spectral Deconvolution and Identification System, NIST). Количественные результаты получали с использованием программы ChemStation G1701DA. Градуировочный график строили по зависимости соотношения площадей морфина, кодеина и дезоморфина к площади внутреннего стандарта (этилморфина) от концентраций морфина, кодеина и дезоморфина. В качестве калибровочных образцов использовали интактную мочу, к которой добавляли спиртовые растворы морфина, кодеина и дезоморфина с известными концентрациями, для морфина – 200, 500, 1000, 10000, 20000 нг/мл, для кодеина – 100, 200, 500, 1000, 4000 нг/мл, дезоморфина – 246, 373, 492, 1230, 2177 нг/мл. Для каждой калибровочной точки проводили по два определения. Пробоподготовку осуществляли по описанной выше схеме анализа. Хромато-масс-спектрометрическое исследование калибровочных образцов проводили в режиме селективного ионного мониторинга.
Характеристические ионы для определения ацетилированных дезоморфина с величинами m/z: 313, 271, 214, кодеина – m/z: 341, 282, 229, морфина – m/z: 327, 369, 310, внутреннего стандарта – m/z: 355, 296. График строили с использованием площадей пиков ионов с величинами m/z 313, 341, 327 и 355. Времена удерживания были следующими: ацетилированный дезоморфин – 12,66 мин (индекс удерживания – RI 2448), ацетилкодеин – 13,43 мин (RI 2610), диацетилморфин – 14,17 мин (RI 2739), ацетилированный этилморфин –13,63 мин (RI 2647). Градуировочные графики линейны в приведенных диапазонах концентраций исследуемых веществ.
При исследовании 1 мкл образца, растворенного в 200 мкл этилацетата, предельно обнаруживаемые концентрации дезоморфина, кодеина, морфина в режиме полного сканирования ионов в интервале масс 45-450 а.е. составили 10 нг/мл.
Обсуждение результатов
Дезоморфин является одной из модификаций структуры морфина. Впервые был получен при поиске заменителей морфина взаимодействием кодеина с тионилхлоридом и последующим восстановлением полученного промежуточного продукта [4]. Не получил широкого распространения в медицинской практике.
Действие дезоморфина очень быстрое, но кратковременное и не сопровождается тошнотой. Использовалось в Швейцарии под коммерческим наименованием permonid [5].
В кустарных условиях дезоморфин получают взаимодействием кодеина, выделенного из лекарственных форм, со смесью кристаллического йода и красного фосфора (рис. 1).
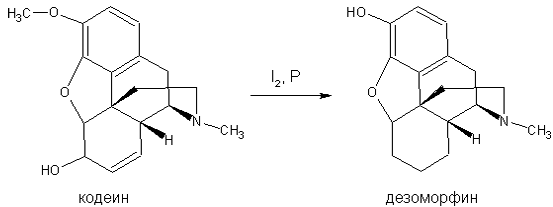
Рис.1. Схема получения дезоморфина из кодеина.
Помимо целевого продукта, в кустарных условиях образуется гамма промежуточных и побочных продуктов, в том числе, идентифицированное встречным синтезом 3-О-метильное производное дезоморфина.
Дезоморфин имеет близкую морфину фармакокинетику и с мочой выводится в основном в виде конъюгатов. При сопоставлении значений концентраций дезоморфина в моче без гидролиза и после кислотного гидролиза видно (таблица 1), что на долю связанных форм приходится от 55 до 100%, в среднем 85% (для 22 образцов).
Таблица 1. Концентрации дезоморфина, кодеина и морфина, найденные в моче потребителей дезоморфина без гидролиза и после кислотного гидролиза.
| № пробы |
Найденная концентрация соединения, нг/мл |
|||||
|
Без гидролиза |
После гидролиза |
|||||
|
ДМ |
К |
М |
ДМ |
К |
М |
|
|
218 |
н.о. |
н.о. |
н.о. |
418 |
следы |
40 |
|
300 |
305 |
297 |
249 |
2709 |
462 |
1616 |
|
361 |
52 |
45 |
31 |
435 |
85 |
164 |
|
501 |
62 |
49 |
22 |
866 |
46 |
137 |
|
1429 |
н.о. |
н.о. |
н.о. |
204 |
следы |
173 |
|
5672 |
н.о. |
следы |
186 |
124 |
24 |
1485 |
|
5995 |
46 |
н.о. |
н.о. |
2718 |
н.о. |
117 |
|
6292 |
2096 |
206 |
342 |
8458 |
232 |
974 |
|
6294 |
н.о. |
следы |
21 |
607 |
н.о. |
94 |
|
6295 |
106 |
следы |
28 |
989 |
34 |
96 |
|
6290 |
299 |
33 |
96 |
1708 |
48 |
246 |
|
6291 |
342 |
45 |
118 |
2234 |
56 |
335 |
|
6940 |
1207 |
н.о. |
н.о. |
9120 |
н.о. |
203 |
|
3220 |
907 |
12252 |
2798 |
2481 |
12982 |
7404 |
|
3844 |
н.и. |
н.и. |
н.и. |
96 |
42 |
245 |
|
6293 |
н.и. |
н.и. |
н.и. |
4643 |
161 |
644 |
|
2569 |
следы |
н.о. |
следы |
691 |
н.о. |
138 |
|
3634 |
54 |
н.о. |
н.о. |
681 |
н.о. |
36 |
|
4044 |
66 |
78 |
н.о. |
148 |
113 |
18 |
|
6343 |
н.и. |
н.и. |
н.и. |
150 |
следы |
177 |
|
6854 |
1327 |
72 |
125 |
5854 |
132 |
596 |
|
6855 |
1136 |
76 |
60 |
8609 |
140 |
314 |
|
6856 |
1108 |
105 |
273 |
7737 |
160 |
902 |
|
6857 |
1913 |
264 |
498 |
5970 |
272 |
1197 |
|
6858 |
858 |
221 |
418 |
2514 |
343 |
1401 |
|
Диапазон |
0-2096 |
0-12252 |
0-2798 |
96-9120 |
0-12982 |
36-7404 |
ДМ – дезоморфин; К – кодеин; М – морфин; н.о. – не обнаружено; н.и. – не исследовалось.
Обладая большей активностью, дезоморфин имеет меньшие действующие концентрации, а выводимые с мочой количества (после кислотного гидролиза) в нашем случае находились в интервале от 96 до 9120 нг/мл. Это значительно ниже определяемых концентраций общего морфина и общего кодеина в моче потребителей героина и кодеина [1]. Данный факт, а также наличие сопутствующих веществ, создает трудности в выявлении дезоморфина при исследовании в тонком слое сорбента. Применение экстракционной очистки после гидролиза мочи позволяет повысить чувствительность тонкослойной хроматографии по отношению к дезоморфину, морфину и кодеину (до 200 – 300 нг/мл по реактиву Фреде). Реактивы Марки и Фреде с дезоморфином дают окраску, идентичную морфину. В таблице 2 приведены величины hRf и RS дезоморфина для системы растворителей толуол – ацетон – этанол – 25% водный раствор аммиака (45:45:7,5:2,5). Спиртовые растворы атропина, антипирина, папаверина исследовали в параллельной хроматографической зоне и проявляли реактивом Драгендорфа в модификации Молдавера. Данные соединения применялись как стандарты в системе общего скрининга для указанной системы растворителей. Как видно из таблицы 2, для определения дезоморфина оптимально использование величины RS относительно кодеина, так как она имеет наименьший коэффициент вариаций (± 6 %).
Таблица 2. Результаты исследования дезоморфина в тонком слое сорбента (n=13).
|
Соединение |
hRf |
RS отн. антипирина |
RS отн. морфина |
RS отн. кодеина |
|
Атропин |
18,8 ± 0,4 |
0,37 ± 0,03 |
- |
- |
|
Антипирин |
52,4 ± 3,0 |
1 |
- |
- |
|
Папаверин |
70,2 ± 3,6 |
1,34 ± 0,03 |
- |
- |
|
Морфин |
20,1 ± 2,4 |
0,38 ± 0,04 |
1 |
0,64 ± 0,07 |
|
Кодеин |
31,5 ± 4,7 |
0,61 ± 0,07 |
1,56 ± 0,15 |
1 |
|
Дезоморфин |
37,0 ± 5,4 |
0,72 ±0,08 |
1,84 ±0,23 |
1,17 ±0,07 |
Сравнение результатов иммунохроматографического анализа и исследования в тонком слое сорбента показало, что в 19 случаях, когда иммунохроматографический тест был положительным, только в 14 случаях методом ТСХ были выявлены опиаты, и лишь в 12 случаях был обнаружен дезоморфин. Таким образом, можно отметить, что при проведении скрининга дезоморфина предпочтение следует отдать иммунным методам анализа и в частности иммунохроматографическому исследованию мочи, как более чувствительному и точному методу [2].
В таблице 3 приведены газо-хроматографические и масс-спектральные характеристики дезоморфина и ряда его производных, которые могут быть использованы для идентификации дезоморфина в объектах исследования. Алкилирование (получение метил- и пентафторбензильных производных дезоморфина) проводили в среде безводного ацетона в присутствии безводного карбоната калия. Триметилсилильные производные получали обработкой дезоморфина БСТФА, содержащего 1% триметилхлорсилана. Ацильные производные были получены действием ангидридов соответствующих кислот на дезоморфин, при этом ацетильные и пропионильные аддукты получали в присутствии безводного пиридина.
Таблица 3. Хромато-масс-спектрометрические характеристики дезоморфина и его производных.
|
Соединение |
[М]+ |
Характеристические ионы m/z, а.е. (интенсивность, %) |
tуд., мин. |
RI |
|
3-Пентафторпропионил- дезоморфин |
417 (100) |
70 (12); 115 (10); 119 (24); 148 (14); 270 (11); 360 (14); 374 (16); 388 (10); 400 (111); 402 (16); 416 (39); 418 (22) |
11,39 |
2151 |
|
3-Трифторацетил- дезоморфин |
367 (100) |
69 (16); 70 (11); 148 (13); 270 (11); 310 (14); 324 (17); 338 (11); 350 (11); 352 (17); 366 (42); 368 (21) |
11,46 |
2166 |
|
Метиловый эфир дезоморфина |
285 (100) |
59 (19); 70 (13); 115 (13); 128 (10); 148 (14); 185 (17); 227 (11); 228 (32); 242 (18); 270 (23); 284 (30); 286 (18) |
12,09 |
2309 |
|
Дезоморфин |
271 (100) |
59 (11); 70 (13); 148 (17); 213 (11); 214 (36); 215 (11); 228 (18); 242 (10); 256 (13); 270 (34); 272 (20) |
12,15 |
2323 |
|
TMС-эфир дезоморфина |
343 (100) |
59 (26); 73 (33); 148 (12); 229 (13); 271 (35); 285 (17); 286 (31); 287 (13); 328 (84); 329 (21); 342 (17); 344 (27) |
12,28 |
2355 |
|
3-Ацетилдезоморфин |
313 (100) |
70 (11); 148 (17); 228 (15); 256 (13); 270 (36); 271 (69); 272 (13); 314 (21) |
12,66 |
2448 |
|
3-Пропионилдезоморфин |
327 (79) |
57 (19); 70 (11); 115 (10); 148 (14); 214 (31); 228 (13); 256 (11); 270 (37); 271 (100); 272 (19); 326 (12); 328 (17) |
13,08 |
2539 |
|
Пентафторбензиловый эфир дезоморфина |
451 (34) |
181 (36); 213 (10); 270 (100); 271 (20); 452 (10) |
14,47 |
2788 |
Так как дезоморфин в кустарных условиях получают из фармацевтических препаратов, содержащих кодеин, дезоморфину сопутствуют фармакологически активные вещества. Вариабельность сопутствующих веществ, обнаруженных в моче потребителей дезоморфина, представлена в таблице 4. Во всех образцах присутствуют метаболиты анальгина, часто встречаются кофеин, димедрол, парацетамол, реже – фенобарбитал.
Таблица 4. Сопутствующие вещества, обнаруженные в моче лиц, употреблявших дезоморфин.
Соединение |
Количество проб |
% от общего количества проб |
|
Никотин, котинин |
25 |
100 |
|
Парацетамол |
14 |
56 |
|
Кофеин |
22 |
88 |
|
Метаболиты анальгина |
25 |
100 |
|
Апоморфин |
13 |
52 |
|
Фенобарбитал |
3 |
12 |
|
Димедрол |
16 |
64 |
|
Изониазид |
1 |
4 |
|
Прометазин |
2 |
8 |
|
Ибупрофен |
2 |
8 |
|
Оксазепам |
1 |
4 |
Таким образом, дезоморфин, являясь структурной модификацией морфина, обладает подобными ему фармакокинетическими свойствами и выводится с мочой, в основном, в конъюгированном виде. Это определяет целесообразность проведения на этапе пробоподготовки для исследования в тонком слое сорбента и методом хромато-масс-спектрометрии кислотного гидролиза, который позволяет повысить их чувствительность за счет разрушения связанных форм дезоморфина.
По причине кустарного изготовления дезоморфина, при исследовании биологических объектов ему сопутствует целый ряд лекарственных веществ, что влечет за собой определенные трудности при применении тонкослойной хроматографии и интерпретации ее результатов. В этой связи, иммунохроматографический анализ приобретает значение как более эффективный метод в скрининге дезоморфина.
Описанные газо-хроматографические и масс-спектральные характеристики дезоморфина и ряда его производных могут быть применены для качественной идентификации последнего. Предложен метод, позволяющий дать количественную оценку содержания дезоморфина, морфина и кодеина в моче, используя метод газовой хроматографии – масс-спектрометрии.
Литература
1. Веселовская Н.В., Коваленко А.Е. Наркотики. – М.:"Триада-Х", 2000.– 206 с.
2. Катаев С.С., Гаранин В.П., Смирнова И.Ю. // Актуальные аспекты судебной медицины. Выпуск VII. Сборник научных работ.- Ижевск: Экспертиза, 2001, с. 62-66.
3. Nordal A. // Bulletin on Narcotics, 1956, Vol. VIII, N.1, P.18-27.
4. Small L. F., Yuen K. C., Eilers L. K. // Journ. Amer. Chem. Soc. 1933, V.55, P.3863
5. Weill P.B., Weiss U.// Bulletin on Narcotics, 1951, Vol. II, N. 2. P. 12-31.