Случай посмертной диагностики синдрома Айерса у ребенка позднего периода новорожденности
/ Рыбалкин Р.В., Святовец Е.В. // Избранные вопросы судебно-медицинской экспертизы. — Хабаровск, 2020 — №19. — С. 104-110.

Рыбалкин Р.В., Святовец Е.В.
КГБУЗ «Бюро судебно-медицинской экспертизы» министерства здравоохранения Хабаровского края (нач. – канд. мед. наук А.В. Нестеров), г. Хабаровск
Синдром Айерса (первичная легочная гипертензия – ПЛГ, идиопатическая гипертрофия правого желудочка сердца, болезнь Ayerza–Arrilaga, легочная болезнь Рейно; облитерирующий аллергический эндоваскулит) – редкое заболевание, характеризующееся совокупностью клинических признаков склероза системы легочной артерии (резкий диффузный цианоз, полицитемия, одышка и высокая легочная гипертензия со значительной гипертрофией правой половины сердца). Впервые синдром описан в 1901 г. аргентинскими терапевтами Ayerza Abel и F.C. Arrilaga. Заболевание встречается крайне редко – примерно у 1–2 на 1 млн популяции. Оно может начаться в любом возрасте,
в том числе и в грудном. Этиология заболевания до настоящего времени неизвестна. Существующие предположения говорят о том, что ПЛГ на сегодняшний день не является болезнью с единой этиологией. Среди возможных причин развития выделяют ВИЧ-инфекцию, портальную гипертензию, коллагенозы, врожденные системно-легочные шунты, заболевания щитовидной железы, врожденные пороки сердца, сопровождающиеся увеличением легочного кровотока, лекарственные препараты (амфетамины, L-триптофан и др.). В последние годы обсуждается роль генетических факторов. Примерно в 7 % случаев ПЛГ – это семейное заболевание, которое наследуется по аутосомно-доминантному типу с неполной пенетрантностью. Риск развития семейной патологии выше у лиц женского пола. Патогенез заболевания также недостаточно изучен. Известно два типа ПЛГ. Первый тип, встречающийся в большинстве случаев, обусловлен резкой гипертрофией, фиброзом, фиброэластозом мышечного слоя легочных артериол. При втором типе, более редком, отмечается рыхлый базофильный клеточный фиброз, приводящий к сужению и облитерации легочных венул. Ранние стадии заболевания, как правило, не распознаются. Основной симптом – одышка. В течение долгого времени она остается единственным проявлением заболевания, поэтому зачастую диагноз ставят поздно, когда появляется выраженное поражение сосудов легких – когда присоединяются цианоз, полицитемия, расширение, правосторонняя гипертрофия и недостаточность правого сердца (синдром «легочного» сердца) с повышенным венозным давлением, при аускультации акцентуация и усиление II тона над ЛА, хлопающий I тон; на ЭКГ – правосторонний тип, правосторонняя гипертрофия сердца вследствие перегрузки, нарушение внутрижелудочковой проводимости, на обзорной рентгенограмме – усиление легочного рисунка в прикорневой зоне, выбухание дуги ЛА. Приведенные выше данные свидетельствуют о сложности постановки диагноза ПЛГ, особенно у детей 1-го года жизни, поскольку единственный клинический признак – одышка – у детей первых месяцев может быть симптомом многих заболеваний (врожденный порок сердца, болезни бронхолегочной системы, инфекции).
В приведенном случае в рамках повторной комиссионной экспертизы были предоставлены медицинские документы и результаты судебно-меди-цинского исследования новорожденного ребенка 2018 года рождения возрастом 27 дней (поздний период новорожденности). Из анамнеза известно, что ребенок родился от седьмой беременности, которая протекала на фоне ОРВИ в 23 недели и маловодия. Мать с отягощенным акушерско-гинекологическим анамнезом, соматически здорова. Роды вторые, срочные в 38 недель, оперативные. Показание к операции: рубец на матке, тазовое предлежание плода, плацентарная недостаточность субкомпенсированная форма. Родился с показателями: масса тела 2 900 г. Длина тела 52 см. Окружность головы 35 см. Шкала Апгар через одну минуту 8 баллов, через 5 минут 9 баллов. Период адаптации протекал физиологически. Находился на грудном вскармливании по требованию, сосал активно, не срыгивал. Докармливался смесью по возрасту, усваивал. Неонатальный скрининг без патологии. Выписан домой в удовлетворительном состоянии под наблюдение участкового педиатра.
На педиатрический участок ребенок поступил 6 января 2019 года с первичным патронажем: на 10-е сутки мать ребенка жалоб не предъявляла, состояние ребенка было удовлетворительным. В медицинской документации отмечено, что ребенок находился на грудном вскармливании, кожные покровы субиктеричные, пуповинная рана чистая, без признаков воспаления. Дыхание в легких пуэрильное, хрипов нет, частота дыхания (далее ЧД) 44 в минуту. Тоны сердца ритмичные, ясные, частота сердечных сокращений (далее ЧСС) 138 ударов в минуту. Неврологический статус без патологических синдромов. За время пребывания дома ребенок получал грудное вскармливание молоком, докорм. На 21-е сутки жизни состоялся актив врача-педиатра на дому, которым состояние ребенка расценено как «ближе к удовлетворительному». Врачом отмечена желтушность. В легких дыхание пуэрильное, хрипов нет. ЧД 40 в минуту. Тоны сердца ясные, ритмичные, ЧСС 120 ударов в минуту. Через четыре дня после посещения педиатра, со слов матери, во время кормления ребенок стал сильно беспокоиться, обмяк, перестал дышать. СМП ребенок был доставлен в стационар. При поступлении состояние ребенка было расценено как крайне тяжелое, дыхание по типу Куссмауля. Тоны сердца глухие – 70 ударов в минуту, установлен диагноз: острый ларингит (?), острая дыхательная недостаточность. По тяжести состояния ребенок был срочно транспортирован в реанимационное отделение. При осмотре реаниматологом: ребенок находился без сознания, адинамичен, кожные покровы бледно-серого цвета с желтушным оттенком. Дыхание самостоятельное, резко затруднено, с жестковатым оттенком, со множеством хрипов по всем полям. Тоны сердца едва прослушивались, глухие, ЧСС практически не определялась. Живот увеличен, стула и мочи при поступлении не было. Проводился мониторинг витальных функций. В дальнейшем утрата сознания, отсутствие пульсации на центральных и периферических артериях. Проведенные реанимационные мероприятия эффекта не дали.
При исследовании трупа экспертом высказывается мнение, что у ребенка имел место выраженный дефицит питания. При массе, долженствующей 3 380 г, масса тела настоящая 3 000 г, что соответствует дефициту 11,2 %. Гипотрофия I степени. Кожный покров трупа бледно-серый с желтоватым оттенком. Вероятно, имела место желтуха грудного вскармливания, обусловленная гипогалактией у матери. В ротовой полости установлена эндотрахеальная трубка № 3 на глубину 11 см. Гортань свободна, слизистая оболочка трахеи и бронхов серовато-розового цвета, в просвете их небольшое количество слизистой мокроты. В просвете пищевода находится дистальный конец эндотрахеальной трубки до уровня средней трети. Вилочковая железа расположена в верхнем средостении с пятнисто-точечными кровоизлияниями под тонкой капсулой темно-красного цвета, массой 4,5 г, размерами 4,0 × 2,0 × 0,5 см. Соотношение массы железы к массе тела равно 0,47, что соответствует выраженной гипоплазии тимуса. При внутреннем исследовании иных патологических изменений, помимо наличия общеасфиктических признаков, не выявлено. Причиной смерти, по мнению эксперта, явилась механическая обтурационная асфиксия в результате дефекта интубации, который обусловил невозможность вентиляции легких и насыщение крови кислородом, приведшую к аноксии. При гистологическом исследовании отмечаются очаговые кровоизлияния под эпикард, в толщу миокарда, под висцеральную плевру, под мягкую мозговую оболочку и в веществе мозга из неизмененных эритроцитов без лейкоцитарной реакции, острая субплевральная и альвеолярная эмфизема легких в сочетании со спазмом бронхов, отек мозга, полнокровие в исследованных органах.
Указанная причина смерти повлекла за собой возникновение уголовного дела и огульное преследование врачей, которые оказывали ребенку медицинскую помощь на всех этапах. Однако, учитывая, что смерть ребенка наступила ранее оказания ему реанимационных мероприятий, а это исключает их участие в танатогенезе, и отсутствие заболевания, приведшего в домашних условиях к остановке дыхания, потребовавшего проведения реанимационных мероприятий с интубацией трахеи, возникла необходимость устранения указанных противоречий, что повлекло проведение повторного исследования материалов дела и медицинских документов.
Во время проведения повторного экспертного исследования в результате анализа представленной документации определено, что в исследуемой клинической ситуации имеется картина острой дыхательной недостаточности с признаками гиповентиляции и гемодинамических нарушений, которые внезапно возникли на фоне относительно стабильного общего состояния ребенка.
С учетом отсутствия в представленных документах сведений о какой-либо патологии со стороны внутренних органов, которая могла бы обусловить подобные проявления (врожденный порок сердца, болезни бронхолегочной системы, системные инфекции), более тщательному рассмотрению подвергнуты результаты секционного исследования, в том числе и микроскопического. С этой целью была выполнена повторная гистологическая экспертиза, в результате которой установлены следующие изменения в легких: просветы альвеол в большинстве полей зрения пусты, на отдельных участках эмфизематозно расширены, очагово содержат десквамированные альвеолоциты с увеличенными ядрами, сосуды межальвеолярных перегородок малокровные, выраженный межуточный отек, перегородки многоклеточные с множеством фибробластов, наличием гистиоцитов, очагово немногочисленных лимфоидных клеток, отмечается сохраненный фетальный тип строения артерий – гипертрофия мышечного слоя, склероз с выраженным сужением просветов конечных разветвлений ветвей легочной артерии, эндотелий утолщенный – «многослойный», набухший, в просвете бронхов десквамированный, отек плевры. Таким образом, имела место персистенция фетальных артерий легких с гипертрофией и сужением просветов (облитерацией) сосудов конечных ветвей легочных артерий на фоне выраженных гемоциркуляторных изменений в виде венозного полнокровия, межуточного отека стромы и паренхимы, очаговых кровоизлияний. Наряду с этим при гистологическом исследовании выявлены выраженные дистрофические изменения кардиомиоцитов с фрагментарной гипертрофией и выраженная гидропическая дистрофия гепатоцитов с очагами баллонной дистрофии. Указанные изменения носили вторичный характер и обусловлены патологией сосудистого русла в легких. Также имелись признаки диспластических пороков развития (недоразвитие отдельных терминальных бронхов, гипоплазия коры надпочечников, тимуса).
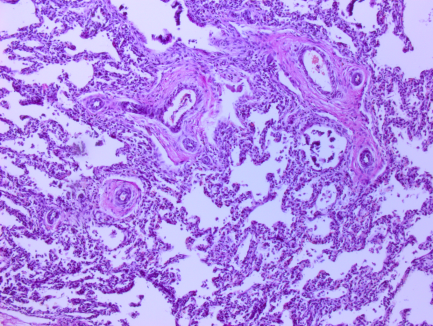
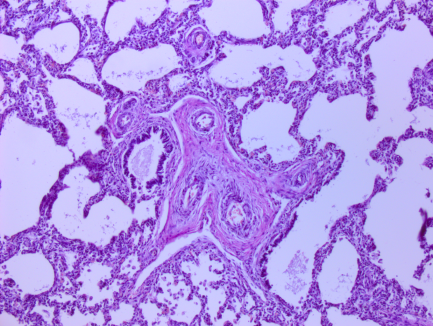
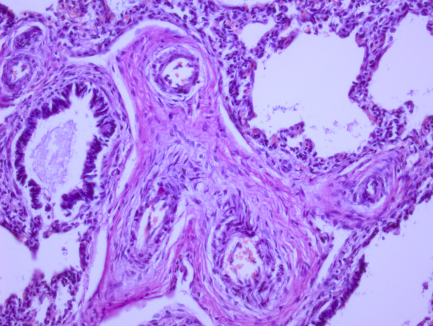
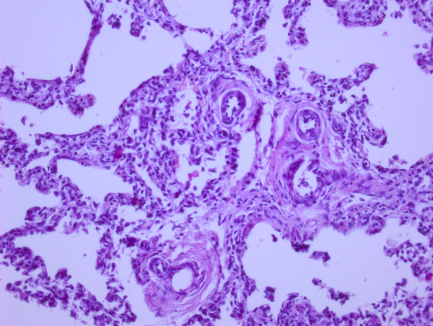
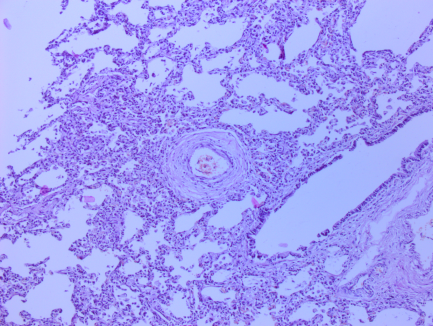
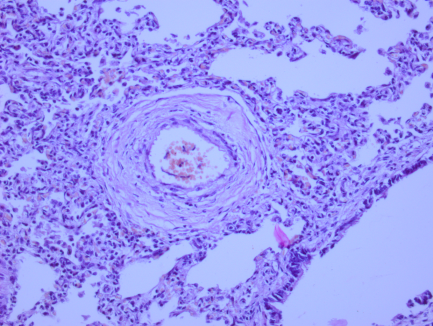
Рис. 1. Микрофотографии изменений в легких (пояснения в тексте)
На всех микрофотографиях различных долей и сегментов легких отмечается резкая гипертрофия, фиброз, фиброэластоз мышечного слоя легочных артериол – признаки первого типа ПЛГ. Ув. 100, 200. Г-Э.
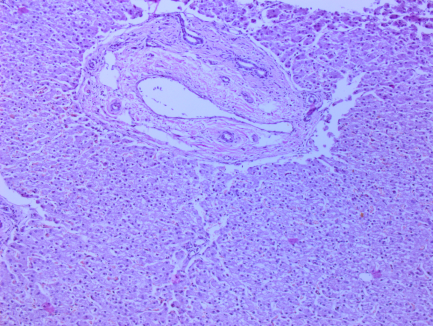
Рис. 2. Печень. Перидуктальный склероз, дистрофия гепатоцитов, очаговая лимофидная инфильтрация портальных трактов. Ув. 100. Окраска гематоксилин-эозин.
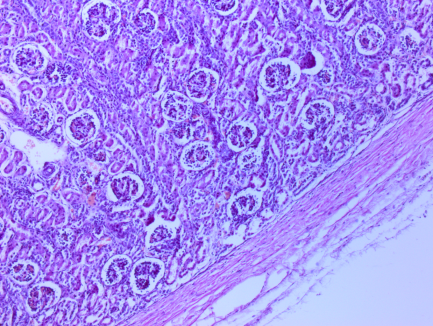
Рис. 3. Почка. Плотно расположенные разного размера клубочки с расширенным мочевым простарнством, отек капсулы. Ув. 100. Окраска гематоксилин-эозин.
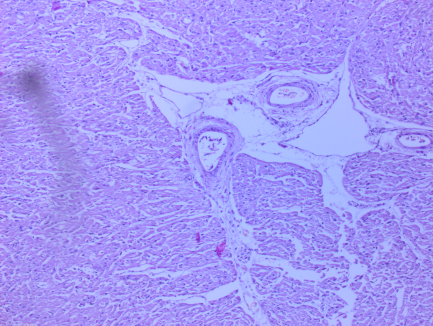
Рис. 4. Сердце. Дистрофия и очаговая гипертрофия кардиомиоицтов, межуточный отек, отек стенок сосудов. Окраска гематоксилин-эозин.
На основании повторного гистологического исследования в совокупности с имеющимися клиническими данными и анамнестическими сведениям экспертная комиссия установила, что в исследуемом случае имела место врожденная первичная легочная гипертензия сосудов малого круга кровообращения, обусловленная персистенцией фетальных артерий легких с гипертрофией и сужением просветов сосудов конечных ветвей легочных артерий.
На основании выявленной патологии с учетом макроморфологических признаков острой, быстро наступившей смерти и диспластических пороков развития экспертная комиссия пришла к выводу о том, что выявленная патология сосудов легких обусловила внезапное нарушение дыхания с развитием острой правожелудочковой недостаточности с быстрым, внезапным развитием нарушения сердечной деятельности и наступлением летального исхода.
Указанный танатогенез подтверждался клинической картиной развития ургентного состояния, результатами гистологического исследования и макроскопическими признаками острой, быстро наступившей смерти, в частности, обнаружением и описанием в протоколе исследования трупа признаков быстро наступившей смерти в виде жидкого состояния крови в полостях сердца и крупных сосудов, обильных сливных трупных пятен, пенистой сукровицы в просвете трахеи и бронхах, очагов острой эмфиземы легких, мелкоточечных кровоизлияний под легочную плевру и эпикард.
Таким образом, кроме определения причины фатальных неблагоприятных последствий, косвенным образом было установлено, что в сложившихся обстоятельствах дефекты проведения интубации в причинной связи с наступившей смертью ребенка не состоят и не могли повлиять на исход ситуации.
Приведенный случай из судебно-медицинской практики интересен редкостью заболевания, сложностью постановки диагноза при секционном и гистологическом исследовании, учитывая отсутствие каких-либо прижизненных проявлений сосудистой патологии и молниеносную форму манифестации. Данный случай иллюстрирует ведущую роль в танатогенезе первичной легочной гипертензии как основной причины летального исхода, обусловленной персистенцией «фетальных» артерий в легких и их гипертрофией.
По нашему мнению, неустановление синдрома Айерса как причины смерти при первичном судебно-медицинском исследовании явилось следствием крайне редкой встречаемости данной патологии (1–2 случая на 1 млн популяции) и соответственно малым количеством встречающейся медицинской информации по данной патологии, в том числе и в патологоанатомической/судебно-медицинской практике. Следует также отметить, что специфика судебно-медицинского исследования трупа, направленная на выявление криминальности в наступлении смерти, накладывает отпечаток на характер диагностического поиска в некоторых случаях в ущерб глубокому этиопатогенетическому анализу и поиску специфических патологических изменений. Во избежание подобных ситуаций, по нашему мнению, судебно-следственным органам следует применять более дифференцированный подход в определении класса проводимого исследования (судебно-медицинского или патологоанатомического) с возможностью в затруднительных случаях совместного исследования и формулирования выводов. Также эффективным было бы регулярное практическое очное обучение специалистов-гистологов в ведущих центрах РФ. В настоящее время такое обучение практически утрачено в угоду действующей программе непрерывного медицинского образования, которое лишает врачей такого обучения, поскольку данная программа характеризуется, по мнению медицинского сообщества, дистанционностью, бессистемностью, мозаичностью приобретения и повторения знаний, отсутствием практической составляющей, коммерциализированностью, отсутствием законодательного утверждения.
похожие статьи
Некомпактный миокард левого желудочка / Голухова Е.З, Шомахов Р.А. — 2013.
Дисплазии соединительной ткани: судебно-медицинская оценка / Конев В.П., Новак В.Г., Сиротин А.А., Шилова М.А. // Матер. IV Всеросс. съезда судебных медиков: тезисы докладов. — Владимир, 1996. — №2. — С. 26-27.
Случай смерти от редкого врожденного порока сердца (синдром “Бланда-Уайта-Гарленда”) / Дмитриева О.А., Голубева А.В., Шегеда М.Г., Баканович И.Б., Артеменко О.В. // Вестник судебной медицины. — Новосибирск, 2016. — №3. — С. 49-53.
Судебно-медицинская экспертиза трупов плодов и новорожденных : Учебное пособие / Витер В.И., Вавилов А.Ю., Бабушкина К.А., Хасанянова С.В. — 2016.
Патология последа / Глуховец Б.И., Глуховец Н.Г. — 2002.
Дефицит витамина К у новорожденного как причина внутрижелудочкового кровоизлияния / Божченко А.П., Грига Э.С. // Избранные вопросы судебно-медицинской экспертизы. — Хабаровск, 2021. — №20. — С. 23-26.
больше материалов в каталогах
Врожденные аномалии и хромосомные нарушения
Беременность, роды, послеродовой и перинатальный период. Смерть детей