Химико-токсикологический анализ лекарственных веществ в крови (плазме, сыворотке) методом высокоэффективной жидкостной хроматографии
/ Крупина Н.А., Краснова Р.Р., Пашовкина Р.Н. // Мат. VI Всеросс. съезда судебных медиков. — М.-Тюмень, 2005.
 методом высокоэффективной жидкостной хроматографии")
(Московская область)
Показана возможность применения метода высокоэффективной жидкостной хроматографии для скрининга и количественного определения лекарственных веществ в крови. Предложены условия изолирования лекарственных веществ из 0,5 мл крови.
Ключевые слова: высокоэффективная жидкостная хроматография, внутренний стандарт, жидкость жидкостная экстракция.
В настоящее время получило распространение злоупотребление медицинскими препаратами и использование лекарственных коктейлей в состав которых входит несколько лекарственных веществ из различных фармакологических групп. Доза каждого из принятых веществ может не достигать токсических концентраций, однако возрастает вероятность случайных и умышленных интоксикаций, нередко приводящих к смертельным исходам в результате фармакологического взаимодействия [1]. В связи с этим резко возрастает значение выбора метода изолирования и методики обнаружения при судебно – химическом исследовании на «неизвестное» вещество. Кроме того, необходимо правильно подобрать объект для исследования, его размерность, идентифицировать экстрагированное вещество с большой чувствительностью, специфичностью и провести количественное определение его. Количественное определение позволяет правильно интерпретировать полученные аналитические результаты и установить, имело ли место в данном случае отравление.
Современная высокоэффективная жидкостная хроматография - один из эффективных методов разделения сложных смесей на отдельные компоненты и проведения качественного и количественного анализа – компонентов разделяемой смеси. В конце 2003 года в лабораторию был приобретен высокоэффективный жидкостной хроматограф Agilent 1100 с диодно – матричным детектором (ДМД). При использовании ДМД проба сканируется каждые несколько миллисекунд, т.е. спектральная информация выдается практически постоянно. Диодная матрица постоянно регистрирует сигналы в ультрафиолетовой и видимой части спектра, обеспечивая таким образом, запись УФ – В - спектров в режиме сканирования. Это позволяет непрерывно снимать при высокой чувствительности неискаженные спектры быстро проходящих через спектральную ячейку компонентов. Данные, полученные от УФ – детектора одновременно на различных длинах волн, обрабатываются и оцениваются с помощью программного обеспечения, которое производит поиск в спектральной библиотеке, выделяет сигнал на определенной длине волны для повышения селективности, вычитает фон и осуществляет другие операции. Программное обеспечение может быть использовано для автоматической сверки УФ – спектров с известными, идентифицировать компоненты и проверять «чистоту» пиков путем сравнения спектров, записанных при начале и окончания выхода каждого пика. Такая автоматизированная система слежения идеальна для скрининга, поскольку позволяет отобрать для дальнейших исследований только «подозрительные» пики.
Целью нашего исследования был подбор оптимальных условий для изолирования лекарственных веществ из крови (плазмы, сыворотки), хроматографического скрининга лекарственных веществ и одновременного количественного определения их с использованием внутреннего стандарта.
Изучив зарубежную литературу по методам изолирования лекарственных веществ из биопроб с дальнейшим исследованием их методом ВЭЖХ [7], мы остановились на методе (несколько модифицировав его), который используется LNS Toxicologie (химико-токсикологическая лаборатория Люксембурга).
Нами также была создана база данных на лекарственные вещества, включающая в себя библиотеку УФ – спектров веществ, их абсолютные и относительные (относительно стандарта) [2 ]времена удерживания.
Экспериментальная часть
- Основа метода. Вещества из крови, сыворотки, плазмы экстрагируются с применением жидкость - жидкостной экстракции смесью н – гексана – дихлорметана – изо – пропанола при рН 9,5. Экстракты анализируются диодно – матричным детектором (ДМД) в градиентном режиме. Обнаружение веществ осуществляется по временам удерживания и УФ спектрам веществ. Количественное определение проводится с внутренним стандартом 5 – (4 – Метилфенил) – 5 – фенилгидантоином (MPPH).
- Объекты исследования. Объектами исследования являются кровь, сыворотка, плазма. Модельные исследования проводились на плазме, не содержащей лекарственных веществ, полученной из отделения переливания крови МОНИКИ им. М.Ф.Владимирского. В качестве реальных объектов использовали кровь, сыворотку живых лиц и кровь трупов, поступающих в отдел на исследование.
- Реактивы. Аммиачный буфер рН 9,5, насыщенный раствор сульфата аммония, н – гексан, дихлорметан, изо – пропанол, ацетонитрил для ВЭЖХ, МРРН, о-фосфорная кислота 85%, однозамещенный фосфат калия (Merck), стандартные растворы веществ (смесь): кофеин, фенобарбитал, димедрол, MPPH, феназепам в концентрации по 10мкг/мл.
- Приборы и принадлежности. Жидкостной хроматограф с диодно матричным детектором Agilent 1100, аппарат для встряхивания (горизонтальный тип встряхивания) 300 колеб./мин, центрифуга «Joan» 4000 об/мин, концентратор экстрактов с термоблоком, РН – тестер Checker, рН – метр (модификации рН 211), водоструйный насос, пипетки переменного объема 100 – 1000 мкл и 10 – 100 мкл с одноразовыми наконечниками, приспособление для фильтрования подвижной фазы.
- Условия обнаружения и определения. Исследование проводили на жидкостном хроматографе Agilent 1100 с диодно матричным детектором в градиентном режиме.
- Колонка:ZORBAX Eclipse XDB-C8 4,6 х 150 мм, 5 мкм;
- Предколонка ZORBAX SB – C18 4,6 х 12,5 мм, 5 мкм
- Температура термостата колонки 300С
- Элюент А: вода деионизированная
- Элюент В: Ацетонитрил ВЭЖХ Градиент
- Элюент Д: Фосфатный буфер рН 3,8
- Градиент:
| Время (мин) | %В | %Д |
| 0 | 15 | 85 |
| 8 | 35 | 65 |
| 17 | 65 | 35 |
| 21 | 65 | 20 |
| 26 | 15 | 20 |
| 7 - 0 | 15 | 85 |
- Скорость потока: 1 мл / мин; от 7 – 0 минут кондиционирование.
- Длина волны: 220 нм. Объем вводимой пробы: 40 мкл.
Стандартные растворы. Стандартные растворы веществ и внутренний стандарт в концентрации 1мг/мл в метаноле, приготовленные весовым методом из стандартов веществ, чистота которых подтверждена методом хромато-масс спектрометрии.
Калибровочные растворы. Калибровочные образцы готовили из стандартных растворов (1мг/мл), с учетом терапевтических, токсических и летальных концентраций веществ в крови (С мг/л) [4]. Концентрации калибровочных растворов и линейность представлены в табл. 1.
Концентрация внутреннего стандарта (MPPH) 10 мкг/мл.
Таблица 1
Концентрация калибровочных растворов и линейность.
| Вещество | С1 | С2 | С3 | С4 | С5 | Линейность (мг/л) |
| Амитриптилин | 0,2 | 0,5 | 1,0 | 2,0 | 5,0 | 0,2 - 5,0 |
| Диазепам | 0,1 | 0,5 | 1,0 | 5,0 | 0,1 - 5,0 | |
| Пропранолол | 0,05 | 0,5 | 2,0 | 5,0 | 0,05 - 5,0 | |
| Феназепам | 0,05 | 0,1 | 0,5 | 1,0 | 2,0 | 0,05 – 2,0 |
| Фенобарбитал | 5,0 | 10,0 | 20,0 | 50,0 | 100,0 | 5,0 – 100,0 |
Калибровка. Прибор с помощью программного обеспечения ChemStation автоматически проводит интегрирование пиков, при этом строится калибровочный график по данным калибровочной таблицы.
0,5 мл бланковой плазмы помещали в стеклянную пробирку на 12 мл, добавляли по 50 мкл (по 2 пробы) каждой концентрации калибровочных растворов веществ, тщательно перемешивали и затем настаивали в течение 5 минут для распределения в плазме. Добавляли 50 мкл МРРН (внутренний стандарт) в концентрации 10мкг/мл, тщательно перемешивали, 0,5 мл насыщенного раствора сульфата аммония, 0,5 мл аммиачного буфера (рН 9,5), Экстрагировали в течение 5 минут на аппарате для встряхивания (300кол/мин) с 5 мл смеси н – гексан – дихлорметан – изопропанол (60:40:2), центрифугировали 3 минуты при 4000 об/мин. Органическую фазу помещали в виалу на 4 мл и испаряли при температуре 400С. Сухой остаток растворяли в 100 мкл подвижной фазы (15% ацетонитрила и 85% фосфатного буфера рН 3,8) и встряхивали в течение нескольких секунд на минишейкере IKA (2000/мин), 40мкл вводили в ВЭЖХ. Параллельно в вышеописанных условиях производили экстракцию и исследование бланковой плазмы.
Анализ исследуемого объекта. 0,5 мл исследуемой крови (сыворотки) помещали в стеклянную пробирку на 12 мл, добавляли 50 мкл внутреннего стандарта МРРН с концентрацией 10 мкг/мл, перемешивали. Далее анализировали аналогично описанному выше. Предварительно вводится стандартная смесь веществ: кофеин, фенобарбитал, димедрол, MPPH, феназепам в концентрации 10мкг/мл.
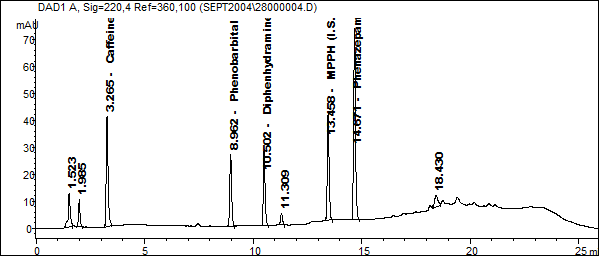
Рис.1. Стандартная смесь веществ (по 80 нг веществ во введённой пробе)
Идентификация лекарственных веществ осуществлялось по временам удерживания и библиотеке УФ – спектров.
Созданная база данных включает в себя следующие лекарственные вещества.
Амидопирин, амитриптилин, аминазин, анальгин, атенолол, атропин, верапамил, галоперидол, грандаксин, диазепам, дибазол, диклофенак, димедрол, карбамазепин, клозапин, кломипрамин, клофелин, кордиамин, кофеин, левомепромазин, левомицетин, миансерин, нитразепам, новокаин, нордазепам, но – шпа, оксазепам, папаверин, парацетамол, пропранолол, тиопентал, тиоридазин, трифтазин, феназепам, фенацетин, фенитоин, фенобарбитал, фтивазид, хлоразепат, хлордиазепоксид, циклобарбитал, циклодол, этаминал. Для некоторых из этих веществ были определены пределы обнаружения, определения,% выхода [6], и проведена статистическая обработка [3, 5].
Результаты
Предложенным методом выход лекарственных веществ основного характера в крови составил от 52,2 до 77%. (амитриптилин при С = 0,01 и 0,05мг/л выход 58 и 54,2% соответственно; димедрол при С = 0,025; 0,2; 10,0мг/л выход 77; 58,3; 70% соответственно; диазепам при С =0,1 и 1,0мг/л выход 54,1 и 53,2% соответственно; пропранолол при С = 0,05; 1,0; 5,0мг/л выход 57,1; 53,4; 70% соответственно; феназепам при С = 0,1; 0,5; 1,0мг/л выход 61,3; 57; 52,2% соответственно. Выход для фенобарбитала при С = 20,0 и 100,0 мг/л составил 16,5 и 15% соответственно. Пределы обнаружения и определения составили соответственно: 10нг/мл; и 50нг/мл (амитриптилин); 20нг/мл и 100 нг/мл (диазепам). 25нг/мл и 100нг/мл (димедрол); 10нг/мл и 50нг/мл (пропранолол); 30нг/мл и 80нг/мл (феназепам); 0,5мкг/мл и 2,0 мкг/мл (фенобарбитал)).
Статистическая обработка результатов (хi - индивидуальный результат, `Х – средняя арифметическая величина; S – стандартное отклонение; RSD (%) – относительное стандартное отклонение; Р – вероятность; Δ`Х – доверительный интервал) представлена в табл. 2.
Таблица 2 Статистическая обработка результатов.
| Наименование веществ |
хi | Метрологические характеристики метода |
| Амитриптилин (С =0,050мг/л) (N = 5) | 0,050 | `Х =0,04 `Х =0,0473 |
| Диазепам (C=0,100 мг/л) (N = 4) |
0,100 | `Х =0,096 S = 0,006 RSD (%) = 6,2 Δ`Х = ± 0,012 Р = 0,99 (`Х ± Δ`Х) = 0,085 – 0,109 |
| Диазепам (C =1,000мг/мл) (N = 5) |
1,000 | `Х=0,947 S = 0,10 RCD (%) =9,5 Δ`Х = ± 0,2 Р = 0,99 (`Х ± Δ`Х) =0,748– 1,148 |
| Пропранолол
(C = 0,050 мг/л) (N =5) | 0,050 | `Х =0,045 S = 0,006 RCD (%) =13 Δ`Х = ± 0,012 Р = 0,99 (`Х ± Δ`Х) = 0,033 – 0,057 |
| Феназепам (С =0,100мг/л) (N =5) |
0,100 | `Х =0,107 S = 0,009 RCD (%) =8,4 Δ`Х = ± 0,018 Р = 0,99 (`Х ± Δ`Х) = 0,089 – 0,125 |
| Феназепам
(С=1,000мг/л) (N = 5) | 1,000 | `Х = 0,995 S = 0,08 RSD (%) =8,0 Δ`Х = ± 0,16 Р = 0,99 (`Х ± Δ`Х) = 0,835 – 1,155 |
| Фенобарбитал (C =10,000мг/л) (N =5) |
10.000 | `Х= 9,462 S = 0,5 RCD (%) =5,3 Δ`Х = ± 0,1 Р = 0,99 (`Х ± Δ`Х) =8,462 – 10,462 |
Описание случаев из практики
По вышеописанной методике произведено количественное определение лекарственных веществ в 50 экспертных случаях. Часть из них представлена в табл.3. Найденные вещества также были подтверждены методом хромато-масс спектрометрии. Приведены хроматограмма экстракта из крови (рис. 1.) и калибровочный график для количественного определения феназепама (рис. 2.), полученных при исследовании случая №2.
Таблица 3 Случаи из практики
№ |
Обстоятельства дела | Найденные вещества (С=мг/л) |
| 1. | Суицид. Поступила из АРО больницы. |
Диазепам (3,48) + кордиамин |
| 2. |
В школе принял таблетки феназепама, принесенные одноклассником, поступил в больницу |
Феназепам (0,08) + кордиамин |
| 3. | Отравление неустановленным веществом | Феназепам (0,62) + морфин + алкоголь |
| 4. | Отравление наркотическим веществом. | Феназепам (0,17) + пропранолол (0,31) + морфин |
| 5. | Обнаружен в подъезде | Амитриптилин (0,23) + морфин + алкоголь. |
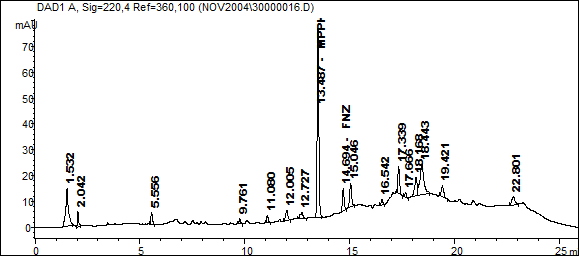
Рис.2. Экстракт из крови.
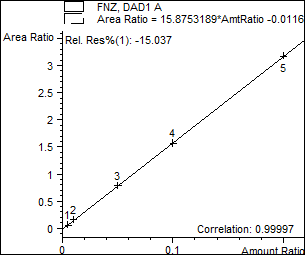
Рис.3.Калибровочныйграфик.(С=0,05мг/л;0,1мг/л;0,5мг/л;1,0мг/л; 2,0мг/л)
Предложенный метод применен в экспертной практике и является специфичным, чувствительным и подходит для рутинных исследований.
Обсуждение результатов
В отечественной литературе, посвященной судебно – химическому исследованию лекарственных веществ в биообъектах недостаточно сведений по скринингу и количественному определению лекарственных веществ в крови с использованием высокоэффективной жидкостной хроматографии. Предложенный метод позволяет в одной пробе провести скрининг лекарственных веществ (при наличии базы данных) и количественное определение.
Пробоподготовка проста и применима для рутинных анализов, требует малых временных затрат. Метод надежен, специфичен, чувствителен и обладает хорошей воспроизводимостью. Использование малой размерности пробы (0,5мл) приводит к сокращению расходов высокотоксичных растворителей.
Выводы
- Предложен метод высокоэффективной жидкостной хроматографии позволяющий в 0,5 мл крови (сыворотки) провести скрининг и количественное определение широкого спектра лекарственных веществ.
- Количественное определение с внутренним стандартом, добавленным в пробу перед экстракцией, позволяет исключить влияние потерь при экстракции и дозировании. В качестве внутреннего стандарта предложено использование химического соединения - 5-(4-метилфенил) –5 – фенилгидантоина, которое не встречается в реальных образцах.
- Метод может быть использован как для терапевтического мониторинга лекарственных веществ, так и для экспресс-диагностики острых отравлений.
- В дальнейшем нами планируются работы по расширению базы данных лекарственных веществ и исследования по пробоподготовке с использованием твёрдо-фазной экстракции.
похожие статьи
Особенности распределения 2,4- и 2,6-ди-трет-бутилгидроксибензола в организме теплокровных животных / Шорманов В.К., Цацуа Е.П., Асташкина А.П. // Судебно-медицинская экспертиза. — М., 2019. — №1. — С. 36-42.